Paired-end sequencing - 97% OTU¶
This tutorial describes a standard micca pipeline for the analysis of overlapping paired-end sequences. The main products of this pipeline are:
- the Operational Taxonomic Units (OTUs), defined clustering the processed sequences at a similarity threshold of 97%;
- an OTU table, containining the number of times each OTU is observed in each sample;
- a taxonomic classification for each OTU;
- an OTU phylogenetic tree.
This pipeline is intended for different platforms, such as Illumina MiSeq and Illumina HiSeq. Although this tutorial explains how to apply the pipeline to 16S rRNA amplicons, it can be adapted to others markers gene/spacers, e.g. Internal Transcribed Spacer (ITS), 18S or 28S.
Table of Contents
Dataset download¶
The following paired-end 16S rRNA dataset contains 34 samples in FASTQ format (V3-V4 region, 341F 5’-CCTACGGGNGGCWGCAG-3’, 806Rmod 5’-GACTACNVGGGTWTCTAATCC-3’).
Samples comes from the paper “Diversity and Cyclical Seasonal Transitions in the Bacterial Community in a Large and Deep Perialpine Lake” were seasonal variations in the bacterioplankton community composition in the lake Garda were analized. Sampling was carried out at monthly intervals in three layers representative of the epilimnetic and euphotic zones of the lake, 1, 10, and 20 m. The dataset contains only a subset of the entire study (2014 samples only) and raw data were randomly subsampled at 3000 sequences per sample.
The 2x300-bp paired-end sequencing was carried out on an Illumina MiSeq.
Open a terminal, download the data and prepare the working directory:
wget ftp://ftp.fmach.it/metagenomics/micca/examples/garda.tar.gz
tar -zxvf garda.tar.gz
cd garda
Merge paired-end sequences¶
Overlapping paired-end sequences must be merged to obtain consensus sequences (sometimes called assembly). This operation can be performed with the mergepairs command.
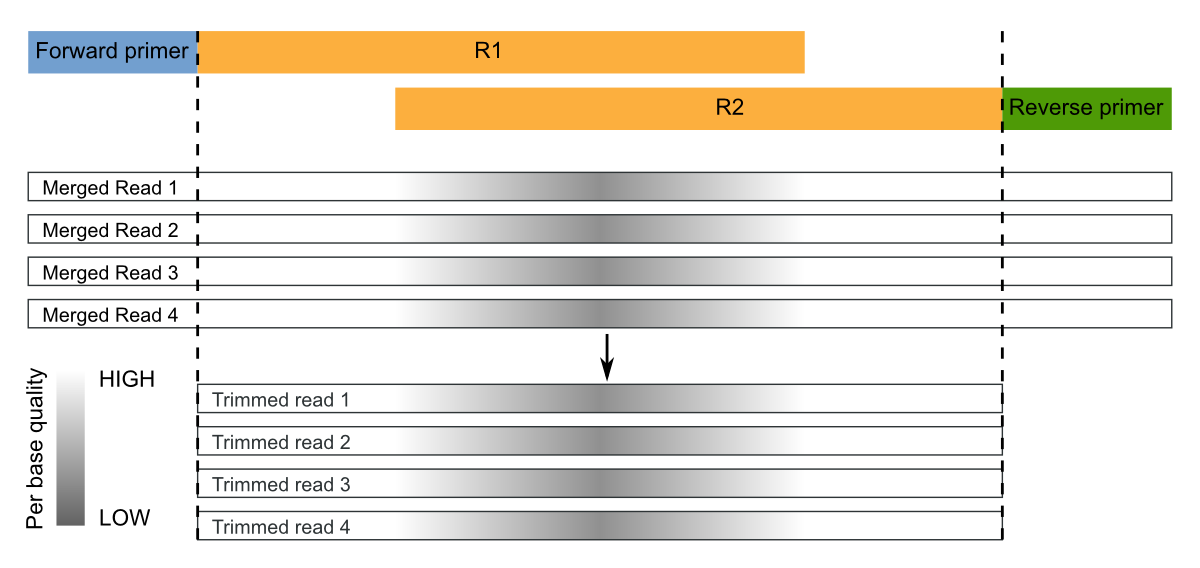
Moreover, the command merges the samples in a single file where sample names are appended to the sequence identifier, as in merge and split commands.
Since the sequenced region is about of 465-bp (806-341) and the reads are of 300-bp, the overlap region is quite large ((2x300)-465=135 bp), as rule of thumb we set a minimum overlap length of 100 and maximum number of allowed mismatches of about 1/3, say 30:
micca mergepairs -i fastq/*_R1*.fastq -o merged.fastq -l 100 -d 30
Note
Starting from micca 1.6.0 staggered read pairs (staggered pairs are pairs
where the 3’ end of the reverse read has an overhang to the left of the 5’
end of the forward read) will be merged by default. To override this feature
(and therefore to discard staggered alignments) set the -n/--nostagger
option.
Note
mergepairs works with FASTQ files only.
Note
Reverse file names will be constructed by replacing the string _R1 in
the forward file name with _R2 (typical in Illumina file names, see
options -p/--pattern and -e/--repl).
Compute reads statistics¶
We can report sequences statistics computed on the file merged.fastq. Run
the command stats:
micca stats -i merged.fastq -o stats_merged
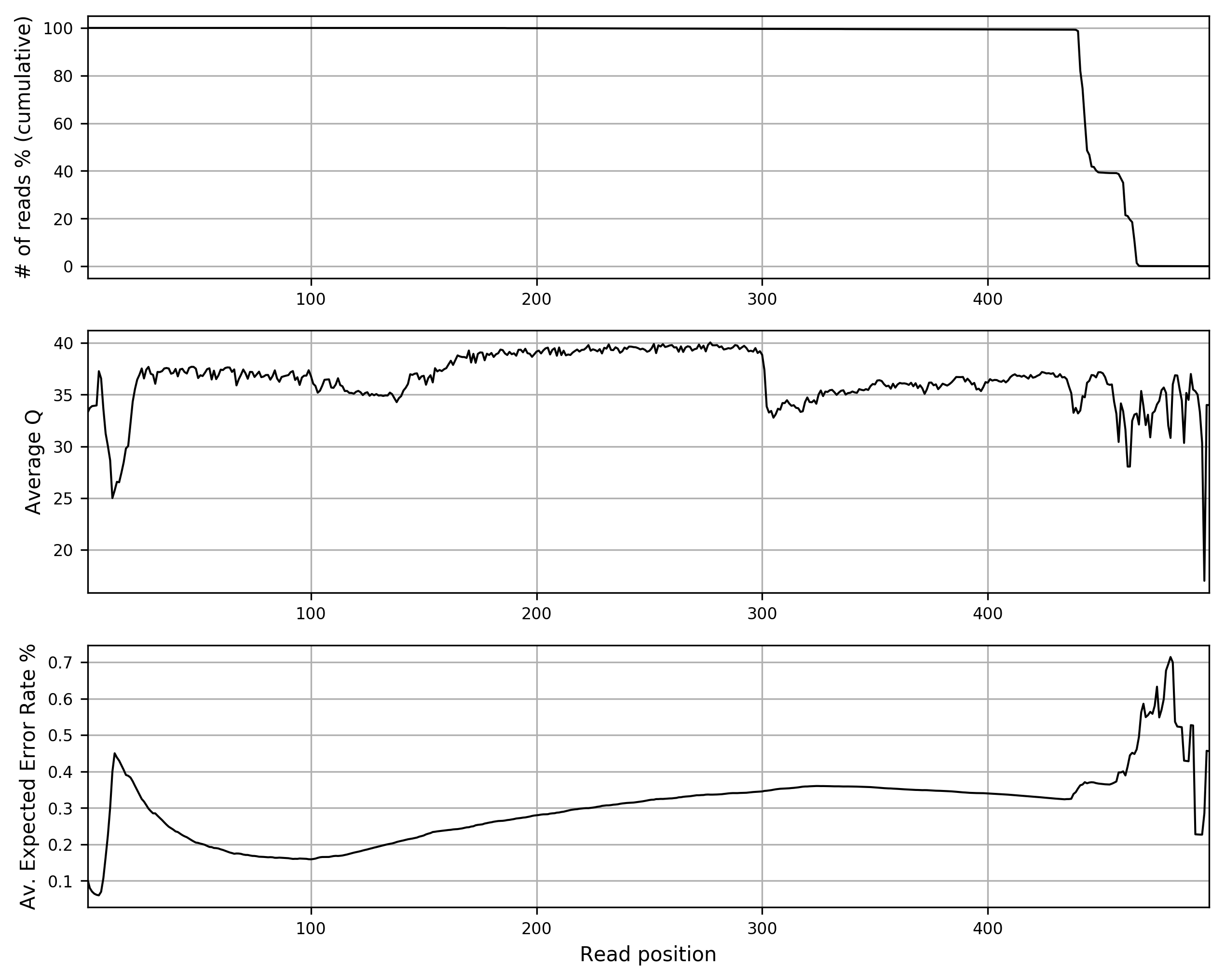
The command reports in 3 text files and in the relative plots (in PNG format) the length distribution, the Q score distribution and a quality summary. The quality summary plot (stats_merged/stats_qualsumm_plot.png) is reported below:
Primer trimming¶
Segments which match PCR primers should be now removed. For paired-end (already merged) reads, we recommend to trim both forward and reverse primers and discard reads that do not contain the forward OR the reverse primer.
These operations can be performed with the trim command:
micca trim -i merged.fastq -o trimmed.fastq -w CCTACGGGNGGCWGCAG -r GACTACNVGGGTWTCTAATCC -W -R -c
The option -W/--duforward and -R/--dureverse ensures that reads that do
not contain the forward or the reverse primer will be discarded. With the option
-c/--searchrc the command searches reverse complement primers too.
Quality filtering¶
Producing high-quality OTUs requires high-quality reads. filter filters sequences according to the maximum allowed expected error (EE) rate %. We recommend values <=1%.
For paired-end reads, we recommend to merge pairs first, then quality filter using a maximum EE threshold with no length truncation.
Warning
Parameters for the filter command should be chosen using the tool filterstats.
Choosing parameters for filtering¶
The command filterstats reports the fraction of reads that would pass for each specified maximum expected error (EE) rate %:
micca filterstats -i trimmed.fastq -o filterstats
Open the PNG file filterstats/stats_plot.png:
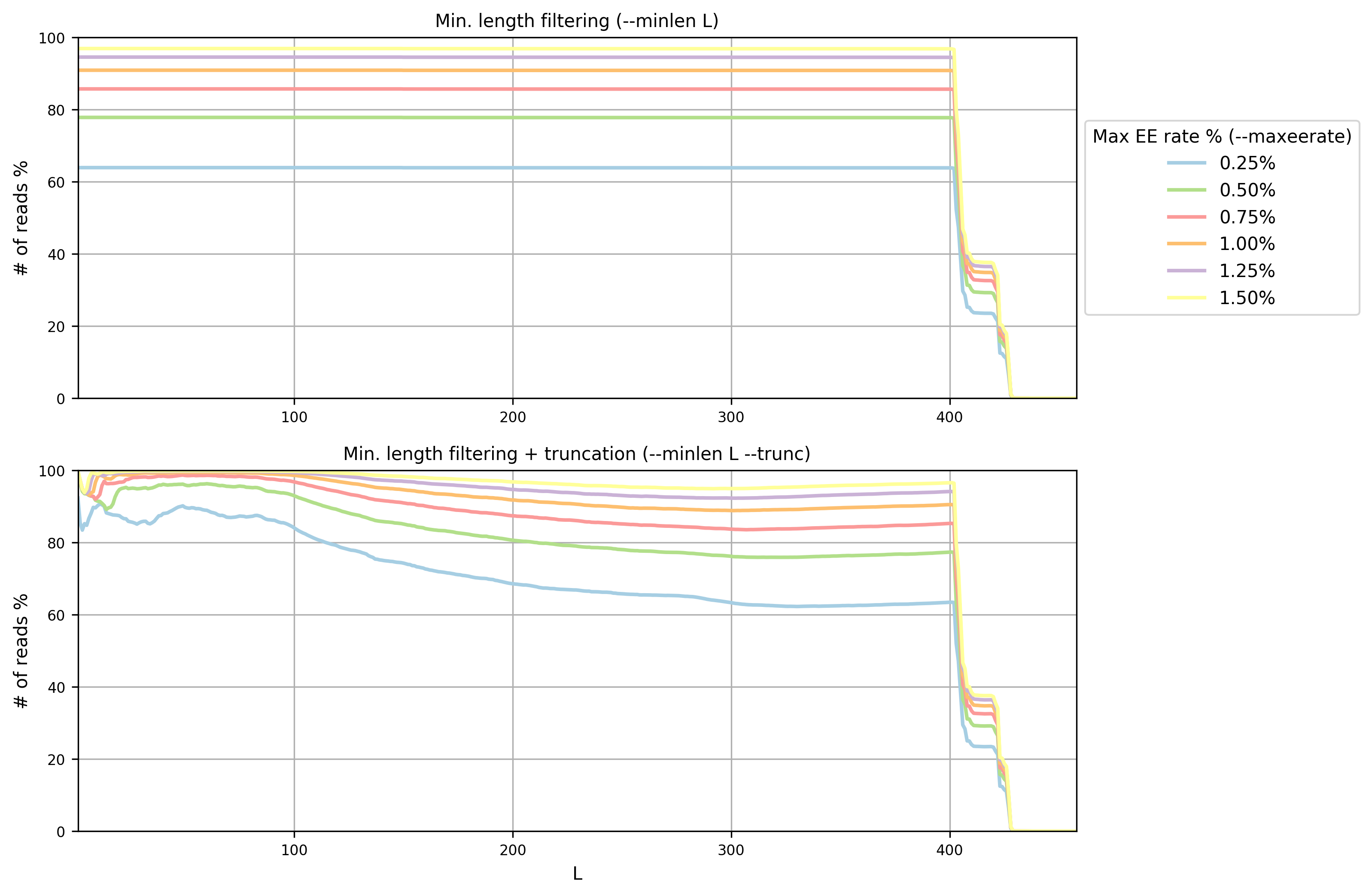
In this case (overlapping paired paired-end reads) we are interested in the plot
on top (minimum length filtering only). A minimum read length (L) of 400 and a
maximum error rate of 0.75% seems to be a good compromise between the
expected error rate and the number of reads remaining. Inspecting the file
filterstats/filterstats_minlen.txt, you can see that more than 85% reads will
pass the filter:
L 0.25 0.5 0.75 1.0 1.25 1.5
...
399 63.856 77.766 85.664 90.844 94.484 96.853
400 63.856 77.765 85.661 90.842 94.481 96.850
401 63.842 77.747 85.643 90.822 94.459 96.827
...
Note
To obtain general sequencing statistics, run the micca command
stats on the file trimmed.fastq.
Filter sequences¶
Now we can run the filter command with the selected parameters:
micca filter -i trimmed.fastq -o filtered.fasta -e 0.75 -m 400
Note
The maximum number of allowed Ns after truncation can be also specified in filterstats and in filter.
OTU picking¶
To characterize the taxonomic structure of the samples, the sequences are now organized into Operational Taxonomic Units (OTUs) at varying levels of identity. An identity of 97% represent the common working definition of bacterial species. The otu command implements several state-of-the-art approaches for OTU clustering, but in this tutorial we will focus on the de novo greedy clustering (see OTU picking and Denoising):
micca otu -m denovo_greedy -i filtered.fasta -o denovo_greedy_otus -t 4 -c
The otu command returns several files in the output directory,
including the SV table (otutable.txt) and a FASTA file containing the
representative sequences (otus.fasta).
Note
See OTU picking and Denoising to see how to apply the de novo swarm, closed-reference and the open-reference OTU picking strategies to these data.
Assign taxonomy¶
Now we can assign taxonomy to each representative sequence using the classify command. In this tutorial we use the RDP (https://doi.org/10.1128/AEM.00062-07) classifier.
Note
See Install on how to install the RDP classifier on your system.
micca classify -m rdp -i denovo_greedy_otus/otus.fasta -o denovo_greedy_otus/taxa.txt
classify returns a taxonomy file like this:
DENOVO1 Bacteria;Cyanobacteria/Chloroplast;Cyanobacteria
DENOVO2 Bacteria;Cyanobacteria/Chloroplast;Cyanobacteria;Family II;Family II;GpIIa
DENOVO3 Bacteria;Chloroflexi;Anaerolineae;Anaerolineales;Anaerolineaceae
DENOVO4 Bacteria;Proteobacteria;Betaproteobacteria;Burkholderiales;Comamonadaceae;Limnohabitans
...
Infer the phylogenetic tree¶
These steps are necessary if you want to use phylogenetic-based metrics such as the UniFrac distance (https://doi.org/10.1128/AEM.01996-06) in the downstream analysis.
Multiple Sequence Alignment (MSA)¶
The msa command provides two approaches for MSA: MUSCLE (https://doi.org/10.1093/nar/gkh340) (de novo alignment) and Nearest Alignment Space Termination (NAST) (https://doi.org/10.1093/nar/gkl244) (which uses a template alignment). In this tutorial we will use the NAST alignment method. For 16S rRNA sequences, a good template alignment is the Greengenes Core Set:
wget ftp://ftp.fmach.it/metagenomics/micca/dbs/core_set.tar.gz
tar -zxvf core_set.tar.gz
At this point we can run the msa command:
micca msa -m nast -i denovo_greedy_otus/otus.fasta -o denovo_greedy_otus/msa.fasta \
--nast-template core_set_aligned.fasta.imputed --nast-threads 4
Build the phylogenetic tree¶
At this point we can build the phylogenetic tree from the MSA using tree:
micca tree -i denovo_greedy_otus/msa.fasta -o denovo_greedy_otus/tree.tree
Note
The output tree is in Newick format.
Midpoint rooting¶
UniFrac metrics require phylogenetic trees to be rooted. The tree can be rooted (in this case at midpoint between the two most distant tips of the tree) using the root command:
micca root -i denovo_greedy_otus/tree.tree -o denovo_greedy_otus/tree_rooted.tree
Note
Tree can also be rooted with the outgroup clade containing selected targets, see root.
Build the BIOM file¶
The Biological Observation Matrix (BIOM) is a common format for representing OTU tables and metadata and is the core data type for downstream analyses in QIIME and in phyloseq. tobiom converts the OTU table and the taxonomy table produced by the previous steps to the BIOM format. In addition, the Sample data can be added:
micca tobiom -i denovo_greedy_otus/otutable.txt -o denovo_greedy_otus/tables.biom \
-t denovo_greedy_otus/taxa.txt -s sampledata.txt