OTU picking and Denoising¶
Usually, amplicon sequences are clustered into Operational Taxonomic Units (OTUs) using a similarity threshold of 97%, which represents the common working definition of bacterial species.
Another approach consists to define the so-called Sequence Variants (SVs, a.k.a Amplicon Sequence Variants - ASVs, Exact Sequence Variants ESVs, zero-radius OTUs - ZOTU, unique sequence variants or sub-OTUs). This approach avoids clustering sequences at a predefined similarity threshold and usually includes a denoising algorithm in order to identify SVs (see UNOISE, DADA2, Deblur, oligotyping and swarm).
The otu command assigns similar sequences (marker genes such as 16S rRNA and the fungal ITS region) to operational taxonomic units or sequence variants (OTUs or SVs).
Methods¶
De novo greedy¶
In denovo greedy clustering (parameter --method denovo_greedy), sequences
are clustered without relying on an external reference database, using an
approach similar to the UPARSE pipeline (https://doi.org/10.1038/nmeth.2604) and
tested in https://doi.org/10.7287/peerj.preprints.1466v1. This protocol
includes in a single command dereplication, clustering and chimera filtering:
- Dereplication. Predict sequence abundances of each sequence by dereplication, order by abundance and discard sequences with abundance value smaller than MINSIZE (option
-s/--minsize, default value 2);- Greedy clustering. Distance (DGC) and abundance-based (AGC) strategies are supported (option
--greedy, see https://doi.org/10.1186/s40168-015-0081-x and https://doi.org/10.7287/peerj.preprints.1466v1 ). Therefore, the candidate representative sequences are obtained;- Chimera filtering (optional). Remove chimeric sequences from the representatives performing a de novo chimera detection (option
--rmchim);- Map sequences. Map sequences to the representatives.
Example:
micca otu -m denovo_greedy -i filtered.fasta -o denovo_greedy_otus -d 0.97 -c -t 4
De novo UNOISE¶
Denoise amplicon sequences using the UNOISE3 protocol. The method is designed for Illumina (paired or unpaired) reads. This protocol includes in a single command dereplication, denoising and chimera filtering:
- Dereplication; Predict sequence abundances of each sequence by dereplication, order by abundance and discard sequences with abundance value smaller than MINSIZE (option
-s/--minsize, default value 8);- Denoising;
- Chimera filtering (optional);
- Map sequences. Map sequences to the representatives.
Example:
micca otu -m denovo_unoise -i filtered.fasta -o denovo_unoise_otus -c -t 4
Closed-reference¶
Sequences are clustered against an external reference database and reads that
could not be matched are discarded (method closed_ref). Example:
Download the reference database (Greengenes), clustered at 97% identity:
wget ftp://ftp.fmach.it/metagenomics/micca/dbs/gg_2013_05.tar.gz
tar -zxvf gg_2013_05.tar.gz
Run the closed-reference protocol:
micca otu -m closed_ref -i filtered.fasta -o closed_ref_otus -r 97_otus.fasta -d 0.97 -t 4
Simply perform a sequence ID matching with the reference taxonomy file (see classify):
cd closed_ref_otus
micca classify -m otuid -i otuids.txt -o taxa.txt -x ../97_otu_taxonomy.txt
Open-reference¶
Open-reference clustering (open_ref): sequences are clustered against an
external reference database (as in Closed-reference) and reads that
could not be matched are clustered with the De novo greedy protocol.
Example:
Download the reference database (Greengenes), clustered at 97% identity:
wget ftp://ftp.fmach.it/metagenomics/micca/dbs/gg_2013_05.tar.gz
tar -zxvf gg_2013_05.tar.gz
Run the open-reference protocol:
micca otu -m open_ref -i filtered.fasta -o open_ref_otus -r 97_otus.fasta -d 0.97 -t 4 -c
Run the VSEARCH-based consensus classifier or the RDP classifier (see classify):
cd open_ref_otus
micca classify -m cons -i otus.fasta -o taxa.txt -r ../97_otus.fasta -x ../97_otu_taxonomy.txt -t 4
De novo swarm¶
In denovo swarm clustering (doi: 10.7717/peerj.593, doi: 10.7717/peerj.1420,
https://github.com/torognes/swarm, parameter --method denovo_swarm),
sequences are clustered without relying on an external reference database. From
https://github.com/torognes/swarm:
The purpose of swarm is to provide a novel clustering algorithm that handles massive sets of amplicons. Results of traditional clustering algorithms are strongly input-order dependent, and rely on an arbitrary global clustering threshold. swarm results are resilient to input-order changes and rely on a small local linking threshold d, representing the maximum number of differences between two amplicons. swarm forms stable, high-resolution clusters, with a high yield of biological information.
otu includes in a single command dereplication, clustering and de novo chimera filtering:
- Dereplication. Predict sequence abundances of each sequence by dereplication, order by abundance and discard sequences with abundance value smaller than MINSIZE (option
--minsizedefault value is 1, i.e. no filtering);- Swarm clustering. Fastidious option is recommended (
--swarm-fastidious);- Chimera filtering (optional).
Warning
Removing ambiguous nucleotides (N) (with the option --maxns 0 in
filter) is mandatory if you use the de novo swarm clustering
method.
Example:
micca filter -i trimmed.fastq -o filtered.fasta -e 0.5 -m 350 -t --maxns 0
micca otu -m denovo_swarm -i filtered.fasta -o otus_denovo_swarm -c --minsize 1 --swarm-fastidious -t 4
Definition of identity¶
In micca, the pairwise identity (except for ‘de novo swarm’ and ‘denovo unoise’) is defined as the edit distance excluding terminal gaps (same as in USEARCH and BLAST):
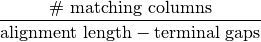
Output files¶
The otu command returns in a single directory 5 files:
- otutable.txt
TAB-delimited file, containing the number of times an OTU is found in each sample (OTU x sample, see Supported file formats):
OTU Mw_01 Mw_02 Mw_03 ... DENOVO1 151 178 177 ... DENOVO2 339 181 142 ... DENOVO3 533 305 63 ... ... ... ... ... ...
- otus.fasta
FASTA containing the representative sequences (OTUs):
>DENOVO1 GACGAACGCTGGCGGCGTGCCTAACACATGCAAGTCGAACGGGG... >DENOVO2 GATGAACGCTAGCTACAGGCTTAACACATGCAAGTCGAGGGGCA... >DENOVO3 AGTGAACGCTGGCGACGTGGTTAAGACATGCAAGTCGAGCGGTA... ...
- otuids.txt
TAB-delimited file which maps the OTU ids to original sequence ids:
DENOVO1 IS0AYJS04JQKIS;sample=Mw_01 DENOVO2 IS0AYJS04JL6RS;sample=Mw_01 DENOVO3 IS0AYJS04H4XNN;sample=Mw_01 ...
- hits.txt
TAB-separated file, three-columns, where each column contains: the matching sequence, the representative (seed) and the identity (if available, see Definition of identity):
IS0AYJS04JE658;sample=Mw_01; IS0AYJS04I4XYN;sample=Mw_01 99.4 IS0AYJS04JPH34;sample=Mw_01; IS0AYJS04JVUBC;sample=Mw_01 98.0 IS0AYJS04I67XN;sample=Mw_01; IS0AYJS04JVUBC;sample=Mw_01 99.7 ...
- otuschim.fasta
- (only for ‘denovo_greedy’, ‘denovo_swarm’ and ‘open_ref’ mathods, when
-c/--rmchimis specified) FASTA file containing the chimeric otus.
Warning
Trimming the sequences to a fixed position before clustering is strongly recommended when they cover partial amplicons or if quality deteriorates towards the end (common when you have long amplicons and single-end sequencing), see Quality filtering.
Note
De novo OTUs are renamed to DENOVO[N] and reference OTUs to REF[N].